Clinical Bioinformatics

Advances in high-throughput experiments such as next-generation sequencing and mass spectrometry gave access to an unprecedented wealth of data to understand systems biology, disease mechanisms and tissue composition. The analysis and interpretation of this data demands sophisticated and efficient methods, tailored to specific research questions. Further, to bring this cutting edge technologies into clinical practice, robust and reproducible workflows are needed to meet the high standards for clinical diagnostics.
Based on single-cell RNA-seq analysis, we want to inform on clinically relevant markers and possible treatment options. We aim at identifying distinct tumor populations, thereby informing on tumor heterogeneity and potential treatment resistant subpopulations. Moreover, the cell type composition of the tumor microenvironment, in particular the presence and variability of immune cell sub populations, can strongly influence treatment response.
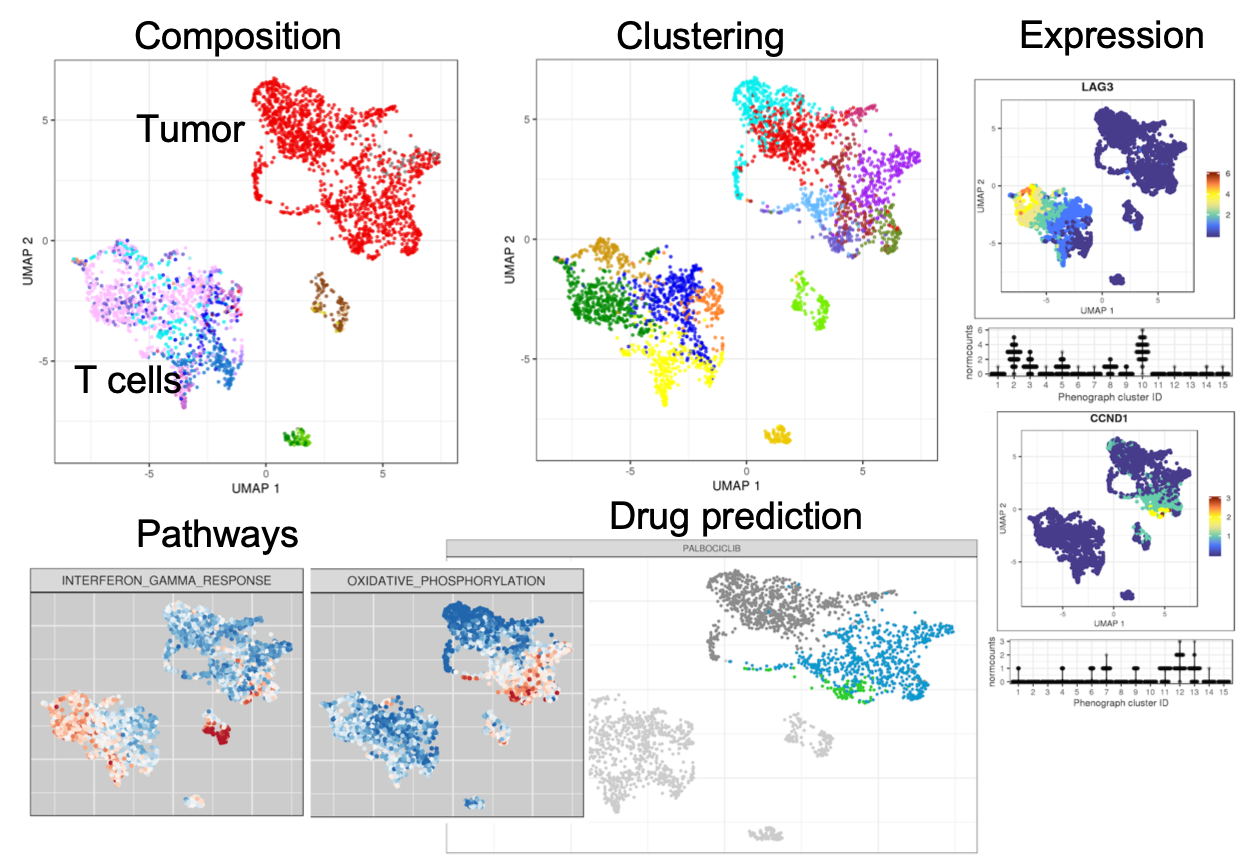
To do so, we designed a workflow to characterize the cell type composition of tumor biopsies based on scRNA-seq data from the 10x Genomics platform. The raw reads are assigned to genes and cells and subsequently filtered and normalized, including several quality control steps. Based on the gene expression profile of each cell, we inform on tumor heterogeneity and immune cell sub populations, and highlight the expression of clinically relevant genes and pathways, as well as perform an in-silico drug prediction.
Types of services
Based on genomic alterations we utilize drug gene interaction databases, literature mining and clinical trials information to enrich genomic information with in-silico based predictions on potential treatment options (e.g., external pageDGIdb, external pageclinicaltrials.gov). These are further enriched with information based on curated databases such as external pageCIViC or external pageOncoKB.
We offer reproducible and scalable pipelines for the analysis of, e.g., gene expression counted based on the external pageICGC standards, or the detection of gene fusion and alternative splicing events that potentially drive disease phenotypes.
Didn't find what you were looking for? We have many other services not listed here for the sake of overview. Furthermore, we regularly include new methods and tools into our portfolio, based on project requests we get from our customers. Please if you did not find the analysis you need and we will see what we can do.
Interested?
If the specific service you are looking for is not listed . There are more services available and we include new ones all the time. You can also have a look at the list of our publications, illustrating the wide scope of applicable services.